
UCLUST algorithm
See also
Dereplication
UCLUST sort order
The UCLUST algorithm divides a set of sequences into clusters. UCLUST is not designed for OTU clustering. See recommended protocols for OTU analysis .
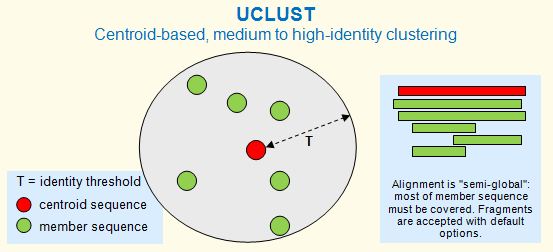
The cluster_fast and cluster_smallmem commands are based on UCLUST. A cluster is defined by one sequence, known as the centroid or representative sequence. Every sequence in the cluster must have similarity above a given identity threshold with the centroid, as shown in the figure below. In previous versions centroids were called seed sequences; this term is no longer used to avoid confusion with alignment seeds (matching words) in algorithms such as BLAST and UBLAST . The identity threshold (T) can be viewed as the radius of a cluster. Clustering commands include cluster_fast and cluister_smallmem .
Recommended identity ranges
UCLUST is effective at identities of ~50% and above for proteins and ~75% and above for nucleotides . At lower identities, this type of method is questionable because (i) alignment quality degrades and (ii) homology cannot be reliably determined from an alignment.
Clustering criteria
UCLUST is designed to find a set of clusters such that:
(1) All centroids have similarity < T to each other, and
(2) All member sequences have similarity >= T to a centroid.
With default parameters, the algorithm is heuristic and condition (1) is not guaranteed to hold, though in practice false negatives (two centroids with similarity >= T) are rare. Note that in general, many different clusterings will satisfy these criteria. For example, a sequence may match two different centroids with identity > T. Ideally, it will be assigned to the closest centroid, but there may be two or more at same distance, in which case the best cluster assignment is ambiguous and an arbitrary choice must be made.
Identities are computed using a global alignment. Clustering based on local alignments could easily be implemented in the USEARCH software, but I believe local clustering is fundamentally flawed . If you have a application that really needs it, I'll add support for local clustering.
Input sequence order
UCLUST is a greedy algorithm , and the order of the input sequences is important. In the cluister_smallmem command, sequences are processed in the order they appear in the input file. If the next sequence matches an existing centroid, it is assigned to that cluster, otherwise it becomes the centroid of a new cluster. This means that sequences should be ordered so that the most appropriate centroids tend to appear earlier in the file. The best order depends on the application; see UCLUST sort order for further discussion. The cluster_fast command can use the input sequence order (this is the default), or can sort by sequence length or by size annotation by specifying -sort length or -sort size,
respectively..
Searching the centroid database
A fundamental step in UCLUST is to compare an input sequence with the centroids identified so far. This is done using the USEARCH algorithm . Most USEARCH parameters, including indexing options , can be used with clustering commands in order to control sensitivity, memory use and speed.
Clustering fragments
If both full-length sequences and fragments are present in the input, then sorting sequences by decreasing length is effective (see sort order , sortbylength ). Fragments usually make poor centroids because two full-length sequences may be similar to a fragment over one segment while being more diverged over the rest of their length. If the fragment is a centroid, this could result in highly divergent full-length sequences being assigned to the same cluster.
OTU clustering
Clustering sequences into Operational Taxonomic Units (OTUs) is a specialized problem. A length sort is not appropriate as this tends to choose centroids that are biological outliers, splitting species and genera into different clusters and inflating alpha diversity. See OTU clustering for a recommended procedure for creating OTUs with USEARCH.
