
OTU benchmark results
See alsoUPARSE home page
OTU / denoising analysis
OTU benchmark methods and data
Introduction
In SSU metagenomics, next-generation reads are clustered into Operational Taxonomic Units (OTUs). This requires quality filtering , dereplication , discarding singletons (optional), and finally clustering into OTUs, typically at a 97% identity threshold.
Benchmark results
The OTU benchmark uses 454 Titanium and Illumina MiSeq reads of Even and Staggered mock communities used for protocol development in the Human Microbiome Project (HMP) . USEARCH results were obtained with the same parameters for all samples. The number of reads per sample ranges from 10,000 (Titanium) to two million (MiSeq). The accuracy of UPARSE was compared to recommended procedures (Sept. 2012) for mothur, QIIME and AmpliconNoise.
| Accuracy measure | Summary | Detailed results (click on image) |
| Sequence quality Are OTUs accurate reconstructions of biological sequences? |
Most USEARCH OTUs are >=99% identical to a biological sequence.
Most QIIME, mothur and AmpliconNoise OTUs are >3% diverged from a biological sequence. Roughly half are chimeric. |
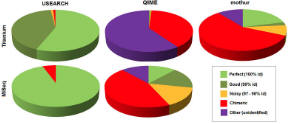 |
| Diversity Does the number of OTUs correspond to the number of species? |
USEARCH generated from 0.8 to 1.0 OTUs per detectable species.
Mothur and AmpliconNoised produced 2.3x to 6.7x more OTUs than species. QIIME produced thousands of OTUs, far more than the number of species. |
|
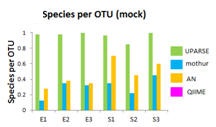
Reference
Edgar, R.C. (2013) UPARSE: Highly accurate OTU sequences from microbial amplicon reads, Nature Methods [ Pubmed:23955772 , dx.doi.org/10.1038/nmeth.2604 ].
