
Pairwise alignment
The -alignpair command aligns two structures
The first structure is given after ‑alignpair , the second structure is specified by the ‑input2 option.
If multiple chains are found in one or both of these files, then an all-vs-all comparison is made to find the highest-scoring pair.
Search output files are supported; only the highest-scoring alignment will be reported.
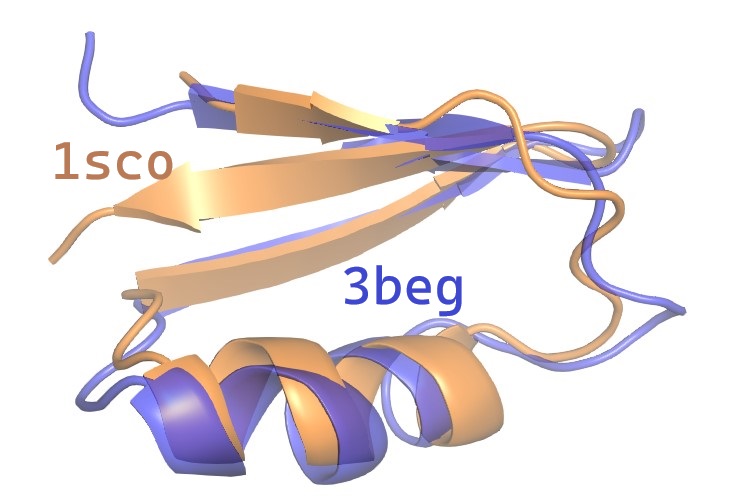
The ‑output option specifies a PDB format file for a rigid rotation of the query chain which optimizes RMSD overlap with the second chain.
You can load the rotated query together with the target chain in a molecure viewer tool such as Pymol to visualize the superposition .
The -alignpair command aligns two structures
The first structure is given after ‑alignpair , the second structure is specified by the ‑input2 option.
If multiple chains are found in one or both of these files, then an all-vs-all comparison is made to find the highest-scoring pair.
Search output files are supported; only the highest-scoring alignment will be reported.
The ‑output option specifies a PDB format file for a rigid rotation of the query chain which optimizes RMSD overlap with the second chain.
You can load the rotated query together with the target chain in a molecure viewer tool such as Pymol to visualize the superposition .
Example
reseek -alignpair 1abc.pdb -input2 2xyz.pdb -aln 1abc_2xyz.txt \
-output 1abc_rotated.pdb
