Lowest common rank probabilities
See also
calc_lcr_probs command
The lowest common rank (LCR) of two sequences is the lowest
rank where both have the same taxon name. For example, Enterococcus avium
and Pilobacter termitis belong to different genera in the
Enterococcaceae family, and their LCR is therefore family.
The identity of a pair of sequences is an approximate
guide to their LCR. For example, if their 16S rRNA identity is 92%, it is a
reasonable guess that their LCR is family. With high confidence, the identity
is too low for them to belong to the same species, and it is almost certain
that the LCR is below phylum. The degree of certainty can be quantified by the
probability that the LCR of a pair of sequences is a particular rank (e.g.,
family) given their pair-wise sequence identity (e.g., 92%).
This probability
depends on how sequences are selected, which can be specified by a frequency
distribution over possible sequences. For a given taxonomy reference database,
the simplest frequency distribution is defined by selecting pairs of sequences at random. However, this distribution usually has strong taxonomic
biases and is likely to be quite different from the distribution encountered
in practice.
This approach confirms that the
conventional 97% OTU threshold is too low.
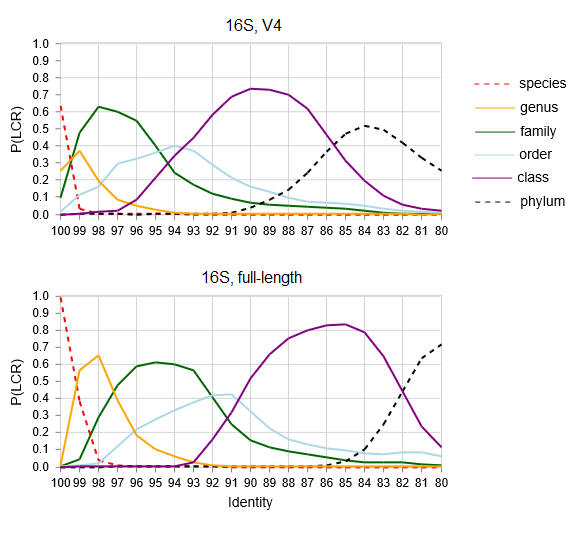
Lowest common rank probability as a function of
identity.
LCR probability for ranks from phylum to species for V4
and full-length 16S sequences.
References (please cite)
R.C. Edgar (2018), Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences, PeerJ 6:e4652
• Cross-validation by identity, novel benchmark strategy enabling realistic accuracy estimates
• Genus accuracy of best methods is 50% on V4 sequences
• Recent algorithms do not improve on RDP Classifier or SINTAX
R.C. Edgar (2018), Taxonomy annotation and guide tree errors in 16S rRNA databases, PeerJ 6:e5030
• Approx. one in five SILVA and Greengenes taxonomy annotations are wrong
• SILVA and Greengenes trees have pervasive conflicts with type strain taxonomies
R.C. Edgar (2017), Updating the 97% identity threshold for 16S ribosomal RNA OTUs, Bioinformatics 34(14) 2371-2375
• Standard 97% OTU identity threshold is too low
• Optimal OTU threshold is 99% for full-length 16S, 100% for V4