
Can SINTAX predict species?
See also
Edgar 2018, "Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences" ( link to paper ).
Shows that V4 accuracy is ~50%, species prediction is not possible, better to use small, authoritative reference.
Edgar 2018, "Taxonomy annotations and guide tree errors in 16S rRNA databases" ( link to paper ).
Shows that annotation error rate of SILVA and Greengenes is ~17%,
On the SINTAX data downloads page you will find a link to a version of the RDP training set with species names. If you use this database with the sintax command , you will usually get a species prediction with a bootstrap confidence. (Not always, because there are species annotations only for the 11,424 of 13,212 sequences where I found a 100% identical match to a named strain).
You should be skeptical of species predictions because it is possible, probably common, to get a high bootstrap value for an incorrect prediction. This happens with genus too, but the problem is worse for species.
If you're using short tags like V4, then it often happens that two or more species have identical tag sequences, making it impossible to identify which species you're looking at. This scenario might not be detectable from the database because the vast majority of species have not been named by taxonomists and do not appear in the RDP training set, so there could be a novel species with an identical sequence. In other words, the reference database is sparse: it has missing data --- lots of missing data.
If you use a "top-hit" classifier like SINTAX or RDP with a sparse reference database, then you get a problem with over-classification as shown in the figure below (taken from the SINTAX paper ). This is what happens: suppose the top hit for your query sequence has 95% identity. Then it probably belongs to a different species. Now suppose the second-best hit has much lower identity, say 90%. The bootstrapping in SINTAX and RDP repeatedly takes a sub-sample of words (8-mers) in the query sequence and checks the top taxonomy when considering only the subset. If there is a big drop in identity between the top hit and second-best hit, then you will get the same top hit every time even if it has relatively low identity, and the result is a high confidence for all ranks in the taxonomy. This is most obvious when the genus is a singleton, i.e. has only one sequence in the database (call it S). Then S is very likely
to be the top hit for any species in that genus, even under sub-sampling of the 8-mers, in which case you'll get a spuriously high confidence that the species of your query is the same as S if it belongs to the same genus. About half (1,157 / 2,472) of the genera in the RDP training set v16 are singletons, so this is a significant concern.
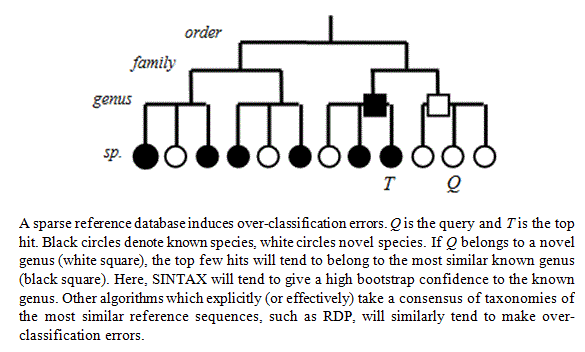
(Figure from the SINTAX paper ).
