Phylogenetic consistency in microbial taxonomy
See also
Microbial taxonomy
Phylogenetic
trees for prokaryotes
The goal of taxonomy has been described as classifying
living organisms into a hierarchy of categories (taxa) that are useful
(based on biologically informative traits) and monophyletic (consistent with
the true phylogenetic tree) (Hennig, 1965), though this point of view is not
universally accepted (Benton, 2000). (See
Edgar 2018, Taxonomy
annotation and guide tree errors in 16S rRNA databases for more
details and references).
With microbial markers such as 16S rRNA, most organisms
are known only from environmental sequencing, and in these cases predictions
of taxonomy must necessarily be made from sequence evidence alone.
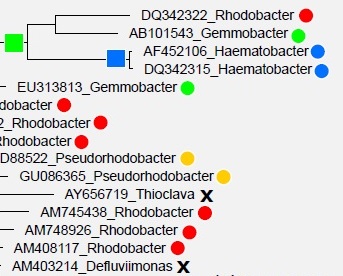 One
approach to predicting taxonomy for environmental sequences and to defining
higher groups for type strains is to infer a tree from 16S rRNA sequences,
on the assumption that the tree will be a reasonable approximation to the
phylogenetic tree. However, in practice this approach is problematic because
of pervasive branching order errors in
estimated trees. This problem has been disregarded or underestimated in
the recent literature.
One
approach to predicting taxonomy for environmental sequences and to defining
higher groups for type strains is to infer a tree from 16S rRNA sequences,
on the assumption that the tree will be a reasonable approximation to the
phylogenetic tree. However, in practice this approach is problematic because
of pervasive branching order errors in
estimated trees. This problem has been disregarded or underestimated in
the recent literature.
Phylogenetic approaches are used by Greengenes and SILVA
for predicting taxonomy of environmental sequences. Bergey's Manual defines
many higher groups based on 16S rRNA similarity, often by construction of a
tree. For example, sub-groups of Firmicutes were constructed by
"...maximum-likelihood analyses of a
dataset comprising about 5000 almost full-length high-quality 16S rRNA
sequences from representatives of the Firmicutes and another 1000
representing the major lines of decent of the three domains Bacteria,
Archaea, and Eucarya. The topology was evaluated by distance matrix and
maximum-parsimony analyses of the dataset". (Bergey's Manual, 2nd ed.,
vol. 3).
The curators of Greengenes, LTP and SILVA have stated
that they consider consistency to be important (emphasis added):
"Greengenes is a dedicated full-length
16S rRNA gene database that provides users with a curated taxonomy
based on de novo tree inference" (McDonald et al., 2012).
"[LTP is based on] comparative sequence
analysis of SSU rRNA [which] has been established as the gold standard for
reconstructing phylogenetic relationships among prokaryotes for
classification purposes" (Yarza et al., 2008).
"SILVA predominantly uses
phylogenetic classification based on an SSU guide tree … discrepancies are
resolved with the overall aim of making classification consistent
with phylogeny" (Yilmaz et al., 2014).
By contrast, taxonomy annotations for environmental
sequences in RDP are predictions made by an algorithm, the
Naive Bayesian Classifier, which does not use a
tree or otherwise explicitly consider phylogeny.