See also
utax command
makeudb_utax command
utax_split command
Taxonomy annotations
Taxonomy training calculates parameters used by the UTAX algorithm to convert a raw score into a confidence value.
Training is performed by the makeudb_utax or utax_train command.
For practical information on training, see How to train UTAX on user data. This page briefly explains how the UTAX training algorithm works.
The training algorithm splits the full reference database into a query subset and a reference subset as shown in the figure below. Taxonomy predictions are made for the query set and compared to the known taxonomies to measure how sensitivity and error rates vary with raw score.
Splitting is performed multiple times at different taxonomic levels. The -utax_splitlevels option specifies the split levels as a string of one-letter codes (d for domain, p for phylum, etc.). Two other pairs of query-reference sets also are used by default: the full reference database as both the query and reference set (indicated by the letter N), and a query database constructed by reversing the sequences in the full reference database (letter V). The 'N' split simulates the case where the query sequence is known, and the 'V' split simulates the case where the query sequence is not homologous to the reference database. In the V case, any predicted taxa are false positives, and in the N case, any incorrect or missing taxonomies are errors. Splitting at the root is indicated by the letter r. A root split (r) should be included if there are two or more top nodes, For example, the Bergey taxonomy used by RDP has two top nodes (d:Bacteria and d:Archaea) so a root split should be included, but k:Fungi is the only top node in the case of the UNITE taxonomy and r should therefore not be used.
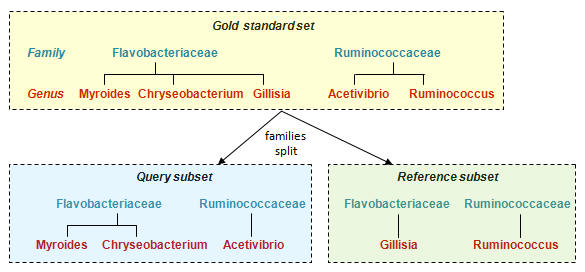
Example: Split at family level
Divide genera for each family into two random subsets, one for the Query subset
and one for the Reference subset. (Discard families with only one genus).
Family is always present in Query and Reference, genus is never present in both.
When trained on the reference subset, a taxonomy classifier could predict family level correctly for all query sequences because the family is known to be present in the training data. If the wrong family is predicted, this is a false positive error, if no family is predicted this is a false negative error. See taxonomy classification errors.
The genus of a query sequence is never present in the
training data, so if a genus is predicted this is an
overclassification error.