See also
fastq_mergepairs command
FASTQ files
Quality scores
Paper describing
merging and filtering
(Edgar & Flyvbjerg, 2015)
Paired read assembler and quality filtering
benchmark results
The process of merging paired reads is sometimes called overlapping or assembly of read pairs. The goal of merging is to convert a pair into a single read containing one sequence and one set of quality scores. A pair must overlap over a significant fraction of its length.
Merging generates a single FASTQ file from FASTQ files for paired forward reads and reverse reads. A pair is merged by aligning the forward read sequence to the reverse-complement of the reverse read sequence. In the overlap region where both reads cover the same bases, a single letter and Q score is derived from the aligned pair of letters and Q scores for each base. If the forward and reverse read agree on the base call, this increases the confidence in the predicted base, increasing the Q score. Conversely, if the reads disagree, this reduces confidence in the base call and decreases the Q score. The adjusted Q scores for matches and mismatches are calculated using Bayesian statistics. The merged Q often exceeds the maximum allowed by the file format, in which case the maximum is used. The maximum Q score for the result of a merge is set by the -fastq_qmaxout option (see FASTQ options for details).
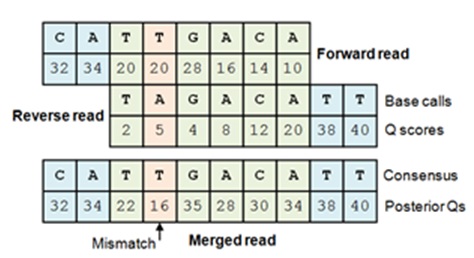
The forward read is aligned to
the reverse-complemented reverse read. If both reads agree on a base call,
the Q score increases due to the increased confidence in the base call per
Eq. 8 in
Edgar & Flyvbjerg 2015. If there is a mismatch, the base call with
higher Q is chosen and the posterior Q score is reduced according to Eq.9.