See also
Read quality filtering
FASTQ format options
Quality scores
Global trimming
If possible, parameters for the fastq_filter command should be chosen manually for each sequencing run by examining the distribution of read length and Phred scores by position in the read, as these characteristics can vary considerably and can have a large impact on downstream analysis. The report given by fastq_eestats2 can be a useful starting point for this exercise. Parameters are chosen to balance conflicting objectives summarized in the table below.
| Objective | Comments | |
| Keep as many reads as possible. |
This is achieved by specifying a short length for
truncation (fastq_trunclen) and a low quality threshold (fastq_truncqual) or
high expected error threshold (fastq_maxee). However, shorter lengths reduce
phylogenetic discrimination, and lower quality
/ higher expected errors will tend to
increase the error rate and can lead to problems such as spurious OTUs. |
|
| Keep as many read positions as possible. | More bases means
better phylogenetic discrimination. However, quality tends to drop towards the
end of an unpaired read, so keeping more bases may increase the number of
errors. With paired reads that overlap, this usually isn't a problem if you
merge the reads using fastq_mergpairs before
filtering. |
|
| Reduce the number of read errors. | Errors are
reduced by (1) truncating the read at a shorter length (if unpaired), and/or (2)
by having a more stringent quality threshold (higher Q or lower
expected errors). However, these will reduce
the total number of bases available for downstream analysis, which may reduce
sensitivity as explained above. |
Filter by minimum quality score or
maximum expected errors?
In the UPARSE paper, I used a minimum quality score (fastq_filter
command with the fastq_truncqual option) to show that a single pipeline with a single set of
parameters gave high accuracy with enormously different input data, from two
million Illumina paired and unpaired reads (forward or reverse) to a few
thousand 454 reads per sample. Given the wide range of input data used in
the UPARSE paper, I could not find a single value of
‑fastq_maxee that worked well with all sets of
reads, and I did not want to give the appearance of tuning (or over-tuning)
to the mock community datasets. However, in practice I would usually
recommend using an expected error filter
as I have found this to be a better predictor of read error rates in most cases.
Paired reads with overlap
If you have paired reads with an overlap that is long enough to merge them
using fastq_mergepairs, then it is usually
best to merge first, then quality filter with
fastq_filter using a maximum expected errors threshold with no length
truncation.
Paired reads with no overlap
If there is no overlap, or the overlap is too short to obtain reliable
alignments, then it is usually best to discard the reverse reads. Typically the
reverse reads have lower quality, and I do not believe the information in the
reverse read can be used effectively (assuming these are amplicon reads). So I
recommend just using the forward reads, i.e. treating them as unpaired (see
next).
Unpaired reads that cover the full
amplicon length
If you have unpaired reads that cover the full length of the amplicons, i.e.
include both primers, then you have two choices: you can truncate at the second
primer (which usually means keeping the full-length reads, unless they extend
further into a non-biological sequence such as an adapter), or you can choose a
shorter length. It may be better to choose a shorter length if the read quality
deteriorates towards the end.
Unpaired reads
that cover partial amplicons
If you have unpaired amplicon reads that do not extend to the second primer,
or have low quality towards the end of the read, then you should truncate to a
fixed length before performing OTU clustering (see
global trimming for an explanation).
Choosing parameters
The simplest way to choose parameters is by trial and error. Set the maximum
expected error parameter (fastq_maxee) to a few reasonable values (say, 0.25,
0.5 and 1), and similarly for the truncation length (say, 50%, 75%, 100% and
125% of the median read length), and measure how many reads are passed by the
filter. The number of reads in a FASTQ file can be determined by using
wc -l filename
to determine the number of lines in the file; divide by 4 to get the number
of reads. The report generated by fastq_stats can
be helpful in guessing suitable parameters.
Example: Analysis of 454 reads
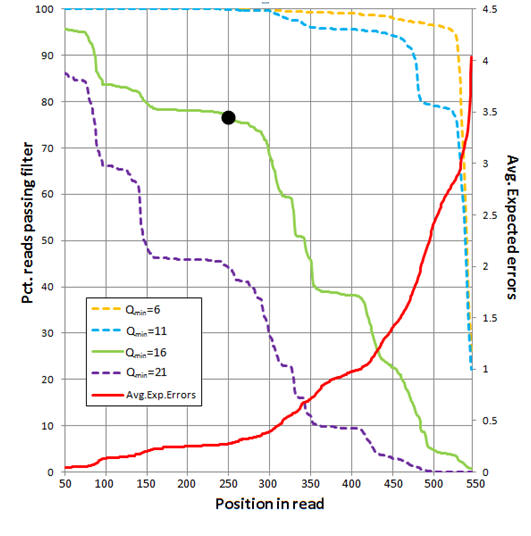
The figure below shows an analysis of 454 reads from the UPARSE paper
(Supplementary Note 3). The average number of expected errors over all reads if
truncated at each position was calculated from the Q scores (red line,
right-hand y axis) using the fastq_stats command.
Four different quality filters are considered with different quality score
thresholds (Qmin, corresponding to one less than the -fastq_truncqual option of
fastq_filter). The fraction of reads passing
each filter if truncated each position is shown (left-hand y axis). The
truncation length L=250 and Qmin=16 values (black dot) were selected as a
compromise between stringent quality filtering to suppress errors (high Q),
keeping as many reads as possible to increase sensitivity to low-abundance
sequences (small L and low Q), keeping as many positions as possible to increase
phylogenetic discrimination (large L), truncating in order to discard
lower-quality regions towards the end of the reads (small L).