Agglomerative clustering is a "bottom-up" method for creating hierarchical clusters. It is implemented in the cluster_agg, cluster_aggd and cluster_edges commands. These commands are highly optimized for speed, often giving performance that is orders of magnitude faster than other packages.
This feature is provided because users sometimes ask for it, though I don't know of a biological application where agglomerative clustering gives better results than the greedy clustering approach used by UCLUST and UPARSE. If you know of a good argument for using an agglomerative method, please let me know.
The algorithm starts by creating one cluster for each input sequence. Then the following step is repeated: identify the closest two clusters and combine them (also called merging, joining or linking). This step is repeated until a single cluster has been formed that contains all the input sequences, or alternatively, if the closest distance between two clusters exceeds a given threshold.
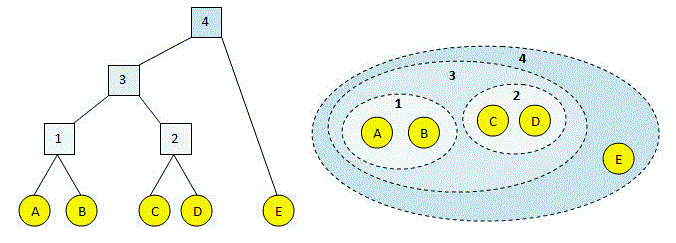
This process is illustrated in the figures below. Letters (in yellow circles) are input sequences. At each step, a new cluster 1, 2 ... is created until all the sequences are combined into a single cluster (#4). In the left-hand figure, the clustering process and result is represented as a binary tree. The children of an internal node are the two clusters that were joined. The right-hand figure shows the same clustering, where dotted lines indicate merged clusters.
Agglomerative clustering requires a definition of linkage, i.e. how to calculate the distance between two clusters in the case where a cluster contains more than one sequence. The most commonly used definitions are minimum distance (single linkage), maximum distance (complete linkage), and average linkage (also called UPGMA). See cluster linkage for details.