See also
fastq_mergepairs command
Introduction to
paired read merging
If the paired read length is longer than the biological sequence in the construct, then the reads overflow into the sequencing primer and adapter sequences which bind the construct to the flow cell as shown below. I call this a "staggered" pair.
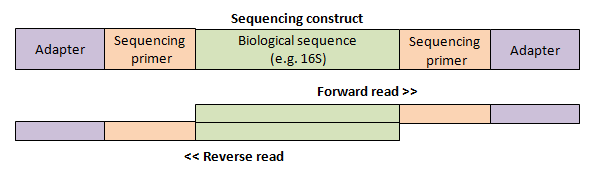
By default, the fastq_mergepairs command trims (discards) any "overhangs" at the ends of the reads which do not align to the other read.
The -fastq_nostagger option causes staggered pairs to
be discarded. This option can be useful when the biological sequence is
known to be longer than the read length, e.g. with 2 x 250 V4 in which case
staggered reads are spurious due to PCR artifacts or other problems. See
also Filtering artifacts by setting a
merge length range.