
Rfam: low-identity nucleotide search benchmark
The Rfam benchmark tests medium- to low-identity nucleotide search. This test should be considered informative rather than rigorous, because rigorous benchmarking of USEARCH against BLAST is not possible.
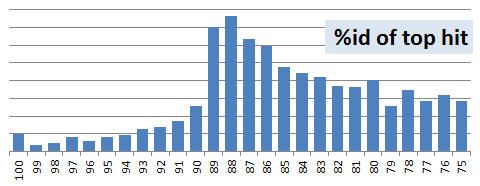
Starting point is Rfam v11.0, which is a 82 Mb FASTA file containing 383,004 RNA sequences. I extracted a subset set containing one randomly-chosen sequence from each Rfam family having > 1 member; the remaining sequences were used as a search database. The query set contains 2,085 sequences. One thousand copies of the subset were concatenated together to make the query set. This was done because search with a single copy of the subset completed in a fraction of a second with USEARCH, making accurate timing measurements impractical. The histogram below shows the distribution of identities between query sequences and the top database hit, according to USEARCH. This shows that the majority of hits have identities in the range 75% - 90%, i.e. low to medium.
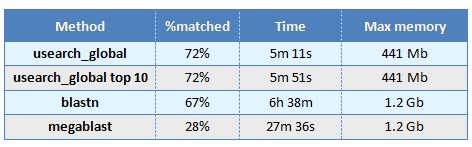
Nucleotide hits with identities < 75% are rarely useful in practice and were therefore excluded. In order to give a rough indication of whether usearch_global and blastn had comparable sensitivity, I took the top hit for each query sequence (by %id for usearch and by E-value for blastn), and discarded the hit unless it had identity at least 75% and covered at least 90% of the query sequence. This number of queries with at least one hit meeting these criteria is reported in the "%matched" column in the above table. Note that megablast has much lower sensitivity than blastn and usearch_global in this case, indicating that usearch_global is significantly faster and significantly more sensitive than megablast.
The "top 10" variant of usearch uses the termination options -maxaccepts 10 -maxrejects 100 to simulate a situation where several top hits are desired rather than a single top hit.
Note that blastn and megablast require more memory than usearch_global in this case. Most likely, this is because of the large number of alignments per query sequence that must be stored in memory on each thread prior to sorting by decreasing E-value and writing to the output file.
See also: hardware configuration.
Command lines
usearch -usearch_global q1000.fa -db rfam.udb -strand plus -id 0.5 -threads 6
-blast6out hits.b6
usearch -usearch_global q1000.fa -db rfam.udb -strand plus -id 0.5 -threads 6 -blast6out hits.b6
blastn -query q1000.fa -db rfam -num_threads 6 -outfmt 6 > hits.b6
blastn -task megablast -query q1000.fa -db rfam -use_index
true -index_name rfam_mb \
-outfmt 6 > hits.b6