| USEARCH manual > UCLUST > recentering |
| recentering |
| See also UCLUST algorithm UCLUST sort order Abundance sort
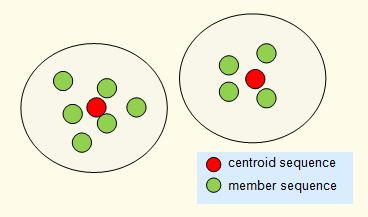
Finding natural or biological centroids
In practice, this is problematic for several reasons. Often, sequences are not neatly separated into groups, so that boundaries are ambiguous. It is difficult (arguably, impossible) to define clustering criteria that always generate clusters that are intuitively natural. This problem has been extensively studied in computer science, see e.g. this Wikipedia article. Recentering
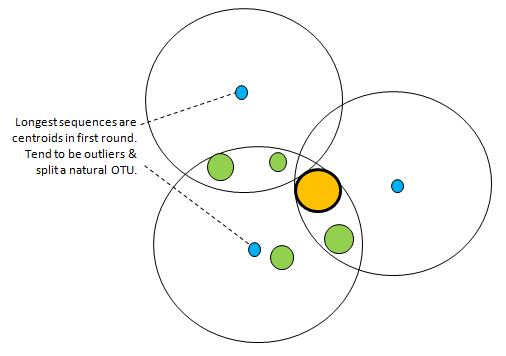
First, UCLUST is run using a length sort (e.g. cluster_fast). This ensures that fragments do not become centroids, but has the problem that the longest sequences (blue dots in figure above). tend to have more errors so tend to be outliers compared to the natural centroids. This tends to split a natural OTU into two or more clusters. This is addressed by constructing consensus sequences for each cluster, which tends to find the dominant sequence in the neighborhood. In the above figure, the dominant sequence is the due to the abundant sequence S indicated by the large orange dot, and the consensus sequences of the lower two clusters will converge on S. The consensus sequence of the top cluster will move closer to the orange dot. Since consensus sequences of neighboring clusters may be identical or closer than the desired distance threshold, a second clustering pass is required to remove this redundancy. An abundance sort is usually preferred for this pass so that biologically correct / biologically more significant sequences are chosen as centroids. After these two passes, the orange sequence above will become the centroid of an OTU which accounts for all the sequences in the figure. In this example, the three OTUs formed by the original outlier sequences are merged after this recentering process. This is typical of what happens in practice with 16S reads: the number of clusters is significantly reduced by recentering. Recentering example usearch -cluster_fast
reads.fasta -consout cons.fasta -id 0.97 -sizeout With these commands, it is not
guaranteed that all the original reads are at least 97% identical to a sequence
in otus.fasta. If desired, this can be addressed by searching the reads against
otus.fasta as a database and clustering any reads that do not match. However, it
is reasonable to discard those reads as they are likely to be spurious (PCR or
sequencing artifacts). |