PILER User Guide
Version 1.0
January 2005
Algorithm developed by Robert C. Edgar and Eugene W. Myers.
Software and manual written by Robert C. Edgar.
This software and documentation is donated to the public domain.
Please visit the web site for requested citation, updates to the software and for information on how to contact the authors for help and feedback.
Introduction
PILER (Parsimonious Inference of a Library of Elementary Repeats) searches a genome sequence for repetitive elements.
Input is a set of one or more DNA sequences. Typically the input is a genome or a subset of a genome such as a chromosome.
PILER search methods
PILER implements four search methods, each designed to find a particular class of repeat. They are summarized in the following table.
|
Type |
Name |
Description |
|
DF |
Dispersed family |
DF search finds families of three or more intact,
isolated copies of a repeat. Dispersed families are most often mobile elements,
but can also be other types such as paralogous exons. "Intact"
means that the copies are globally alignable to each other, isolated means
that they are surrounded by unique sequence (i.e., sequence that has no local
alignments). |
|
TA |
Tandem array |
TA search finds tandem arrays. A tandem array is
a contiguous series of three or more copies of a motif. The copies are
globally alignable to each other within the chosen thresholds for local
alignment. |
|
PS |
Pseudo-satellite |
PS search finds pseudo-satellites. A
pseudo-satellite is a dispersed family whose members cluster in the genome
(i.e., are found close together), but not so close that they form tandem
arrays. |
|
TR |
Terminal repeat |
TR search finds similar pairs that may be
terminal repeats in mobile elements such as the LTR superfamilies or Tc1. |
Software packages
Three software separate packages are required: PILER, PALS and MUSCLE. PALS is used to find local alignments of a genome to itself. MUSCLE is used to create multiple alignments of each repeat family. Expert users can substitute other local and multiple aligners to replace PALS and MUSCLE if desired. The home pages for these packages are:
Using an alternative local aligner
We recommend using the PALS aligner. Local alignments produced by other methods, e.g. BLAST, may be used by converting their output to PALS GFF format. (See the PALS User Guide). The alignment score and maxe fields provided by PALS are not used by PILER so arbitrary placeholder values can be used.
Terminology
A hit is a local alignment of the genome to itself. The two regions aligned by a hit are called images. Given an image in a hit, the other image to which it is aligned is called its partner image. The copy count of a base in the genome is the number of images that cover the base. A pile is a maximal contiguous region of bases with copy count > 0.
PILER-DF
DF (Dispersed Family) search finds families of three or more
intact, isolated copies of a repeat. Dispersed families are most often mobile
elements, but can also be other types such as paralogous exons. "Intact"
means that the copies are globally alignable to each other, isolated means that
they are surrounded by unique sequence (i.e., sequence that has no local
alignments).
Output from PILER-DF is:
(1) An annotation of the input sequences giving locations of intact, isolated copies of repeated elements. These are typically mobile elements, but are sometimes other features such as paralogous exons or microsatellites. Each location is assigned to a family of similar elements that are mutually globally alignable to each other.
(2) A library containing one consensus sequence for each family. This library can be used by another application, e.g. BLAST or RepeatMasker, to find other instances of the repeats in each family and mask them if desired.
The algorithm has three main stages, summarized in the following table.
|
Stage |
Description |
|
Find local alignments. |
The PALS aligner is used to align the genome to
itself. |
|
Transposed repeat signature search. |
TRS search finds families of three or more
intact, isolated copies of a repeat. |
|
Library construction. |
For each family, sequences are extracted and a
multiple alignment built using MUSCLE. A consensus sequence for each family
is constructed from the multiple alignment. |
PILER-DF families and superfamilies
A family is a set of repetitive regions in the genome that are globally alignable to each other. Typically a family is a set of copies of a single type of mobile element. PILER assigns an identifier to each family. The family identifier is two integers separated by a period, e.g. 12.34. The first integer is the "superfamily" index, the second integer is the family index. The family index is 0, 1, 2 .. up to the number of families found by TRS search. If two families have a local alignment to each other in the hit file, they are assigned to the same superfamily. Similarly the superfamily index is 0, 1, 2 .. up to the number of superfamilies found. The superfamily index is provided as a hint to post-PILER analysis. Often, two families in the same superfamily are the same mobile element, or one is a fragment of the other.
DF Stage 1: local alignments
The first stage in PILER-DF creates a single GFF file in PALS format containing a set of local alignments of a genome to itself. The format of this file is described in the PALS User Guide. Note that this is an asymmetrical hit file, meaning that if a hit from region A to region B is included, then hit B to A should not be included. Trivial alignments of a region to itself should also be excluded. Using the ‑self option in PALS produces an output file in the correct format. If the genome is too large to align the entire sequence to itself, then create the hit file as follows.
1. Split the genome into chunks small enough for PALS. E.g., into chromosomes.
2a. Align each chunk to itself using the ‑self option of PALS.
2b. Align each different pair of chunks to each other using the ‑query and ‑target options of PALS. It makes little difference which chunk is chosen to be the target, except that shorter targets may reduce memory requirements.
3. Concatenate the hit files produced in steps 2a and 2b into a single hit file.
DF Stage 2: TRS search
TRS search requires a hit file as input and produces a TRS file as output. The TRS file is in GFF format; it contains the coordinates and family number of each intact, isolated repeat identified by the search.
("TRS" is an abbreviation for "transposed repeat signature", which was chosen for historical reasons. "Dispersed family" is a more accurate name than "transposed repeat" for the kinds of things found by this).
This is done using the piler command with the following options:
piler
-trs pals.gff -out trs.gff [-piles piles.gff]
[-images images.gff] [-famsize
f] [-maxlengthdiffpct
d]
[-multihit]
The file names shown (pals.gff, trs.gff, piles.gff, images.gff) are examples and may be changed as desired. Directory names may be included, e.g.
piler
-trs /genomes/hits/ecoli_self.gff
-out /piler/ecoli/trs.gff
Mandatory parameters are ‑trs, which specifies the hit file, and ‑out, which specifies the TRS output file.
Optional parameters are:
-piles filename
Specifies the name of a GFF file to contain coordinates of each pile identified by TRS search.
-images filename
Specifies the name of a GFF file to contain coordinates of each image from the hit file, annotated with the pile number of the image and its partner image.
‑famsize f
Specifies the minimum size f of a family. The default is the minimum value 3. A value of 1 or 2 will probably produce a large number of false positives. Larger values will increase specificity (reduce the number of false positives) but reduce sensitivity (find fewer mobile element families).
-maxlengthdiffpct d
Specifies the maximum length difference d allowed between two piles in order for them to be considered globally alignable, expressed as a percentage. The default is 5 (i.e, 5%). Larger values will increase sensitivity but may increase the number of false positives. The value may be specified using integer, fixed point or scientific notation (it is parsed using atof).
-multihit
Specifies that multiple images may be used to determine global alignability. By default, a single image is required to span both piles. Allowing multiple images tends to increase sensitivity, but may produce more false positives.
DF Stage 3: Library construction
Library construction takes the genome sequence and TRS output file as input, and produces a library of consensus sequences as output.
This is done using the following steps.
3a. A FASTA file is created for each family.
3b. A multiple alignment is created for each family.
3c. A consensus sequence is created from each multiple alignment.
3d. The consensus sequences are concatenated to create the library.
3a. Making a family FASTA file
Family FASTA files are produced using the piler command with the following options:
piler
-trs2fasta trs.gff -seq genome.fasta [-path fams] [-maxfam m]
[-prefix p]
File names trs.gff and genome.fasta are examples and may be changed as desired.
Mandatory parameters are ‑trs, which specifies the TRS output file, and ‑seq, which specifies a FASTA file containing the genome sequence.
Optional parameters are:
-path fams
The directory where the family FASTA files should be created. Default is the current directory.
-maxfam m
Specifies the maximum number of sequences m to include in a family FASTA file. Too many sequences may exceed the capacity of a multiple aligner, and may create a low-quality consensus sequence. The default is 16.
-prefix p
Specifies a prefix for the PILER family identifier. This is typically an abbreviation for a species or strain, so that family identifiers are unique across several genomes. For example, you might use ‑prefix dmel for D. melanogaster. If so, the FASTA file name for family 12.34 will be dmel.12.34.
3b. Making a family alignment
A family FASTA file can be aligned with MUSCLE, or any other suitable multiple aligner. We recommend MUSCLE with the following parameters:
muscle -in 12.34.fasta -out 12.34_aligned.fasta -maxiters 1 -diags1
Note that there is a space in ‑maxiters 1, but no space in ‑diags1. These options are fast and accurate for closely related DNA sequences.
3c. Making a family consensus sequence
A consensus sequence is produced from a multiple alignment using the piler command with the following options.
piler -cons 12.34_aligned.fasta
-out 12.34_cons.fasta -label 12.34
All options are mandatory: ‑cons specifies the multiple alignment file, ‑out the FASTA file to contain the consensus sequence, and ‑label the sequence name to appear in the FASTA annotation line (following a ">" character).
3d. Concatenating consensus sequences
This is a simple step that can be easily done with standard tools. E.g., in Unix you can use the cat command:
cat *_aligned.fasta > dmel_library.fasta
PILER-DF implementation
To implement the full PILER-DF procedure, you will probably choose to write one or more scripts. The details will vary depending on the chosen script language (e.g., shell) and on the genome(s) to be analyzed. Some experimentation will probably be needed to determine the appropriate details given the length of the genome, available memory and other factors. Also, different parameters are optimal for different genomes, depending on the repeat content. As PILER is a specialized tool for expert users, I don't expect a heavy tech support burden and will probably be happy to discuss your particular application. Please visit the PILER web site /piler to find out how best to contact me.
As an example to get you started, here is a simple script
for the bash shell that implements
PILER-DF with default parameters for a genome short enough to be aligned in a
single chunk. The genome must be in a file genome.fasta in the current
directory. The library is written to piler_library.fasta.
pals -self genome.fasta hit.gff
piler -trs
hit.gff -out trs.gff
mkdir fams
piler -trs2fasta trs.gff -seq genome.fasta
-path fams
mkdir aligned_fams
cd fams
for fam in *
do
muscle -in $fam -out ../aligned_fams/$fam -maxiters 1 -diags1
done
cd ..
mkdir cons
cd aligned_fams
for fam in *
do
piler -cons $fam
-out ../cons/$fam -label $fam
done
cd ../cons
cat * > ../piler_library.fasta
Comparison with other annotation
It is often useful to compare PILER annotations with those from other programs, such as RepeatMasker. The ‑annot option of the piler command generates a simple visual summary of a third party annotation for a given region in the genome, and appends the summary to each record in a GFF file. Here is an example summary:
---aaaaaBbbbbbbbbbbB ATREP13(35%) ATREP7(100%)
The summary starts with a 16-character "picture" that represents the region between the Start and End coordinates given in the GFF record. The picture is fixed-width, so the scale varies for each record; i.e., a single character in the picture may represent a different number of bases in different pictures. Characters are letters, representing a repeat annotation, or a dash (–) indicating that there is no annotation for those bases. A letter a, b, .. refers to a repeat name following the picture. Letters map to repeats in the obvious way: a means the first repeat mentioned, b the second, and so on. If repeat annotations overlap (which never happens with RepeatMasker), then the last-mentioned repeat wins. The repeat is followed by a percentage that indicates the fraction of the full-length repeat matching the region. A value of 100% indicates a full-length repeat, < 100% indicates a fragment. No percentage is given for repeat classes that vary in length, such as "simple repeats" (microsatellites) and low complexity regions (e.g, AT_rich). An upper case letter represents the endpoint of the library sequence. This is a useful indicator for fragments, where one end of the alignment could be the true endpoint of the repeat. For obvious reasons, upper case letters are given only for fixed-length repeat classes, such as mobile elements, and not for simple repeats or low complexity regions. If the GFF Strand field is –, indicating that the match is reverse complemented, then the picture is reversed.
Pictures are created using the -annot option of the piler command as follows:
piler -annot
input.gff -rep repeats.gff
-out output.gff
The parameters are all mandatory: -annot specifies the GFF file to be annotated, ‑rep a GFF file containing third party annotation, and ‑out the file to contain the updated input file.
The third party annotation file is in GFF format with the following fields. The Feature field must be set to repeat (other records are ignored). The Attributes field must be formatted as in the following example:
Repeat HETRP_DM Satellite 1518 1650 19
The attribute starts with Repeat, and is followed by five fields separated by exactly one space, as specified in the following table.
|
Field |
Example above |
Description |
|
Name |
HETRP_RM |
A short symbolic name
for the repeat. |
|
Class |
Satellite |
A short symbolic name
for a class of repeats. |
|
Start |
1518 |
Start position in the
repeat (1 is the first base). |
|
End |
1650 |
End position in the
repeat. |
|
Left |
19 |
Number of bases in the repeat following End. |
In the above table, "the repeat" refers to a library
sequence that aligns to the genome, for example a RepBase sequence in a RepeatMasker
library. The Start, End and Left fields are defined by positions in that library sequence. Note
that End + Left is the length of the
library sequence. If the repeat class does not have a fixed length, e.g.
microsatellites or low complexity regions, Start,
End and Left should appear as a dot (.).
The Python script rm2gff.py converts a RepeatMasker .out file to .gff format. Here is an example record.
chr1 RepeatMasker repeat 18331 18642 1202 - .
Repeat ATHATN7 DNA/HAT 311 598 0
Here are attributes from three trs records (produced from a PILER-DF search of Arabidopsis) with annotation pictures added using -annot:
Family
31.24 ; Pile 1359 ; Annot
"AaaaaaaaaaaaaaaaaaaA ATREP2A(100%)"
Family
48.37 ; Pile 1343 ; Annot
"---aaaaaBbbbbbbbbbbB ATREP13(64%) ATREP7(100%)"
Family
60.44 ; Pile 1346 ; Annot
"--------------------"
The pictures show that the first record is an excellent match to the ATREP2A repeat. The second match appears to contain a full-length repeat (ATREP7), a fragment of a second type of repeat (ATREP13), plus a short region that is unannotated by RepeatMasker. Family 48.37 is therefore a questionable prediction. The third record is a region that is entirely unannotated by RepeatMasker, so is a novel repeat (i.e., does not match RepBase).
TRS Report
The Python script trs_report.py creates a readable report from an (optionally annotated)
PILER-DF (also called TRS) output file. We find this report helpful in
reviewing and evaluating PILER results. The report sorts TRS records first by
superfamily and then by family. The sequence, coordinates, strand and length of
each family member is given, together with the pile number and annotation. The
number of piles (family size) and average length are given in at the end of
each family. Following are some typical Arabidopsis
families.
Family 0.8
Sequence
Start End +
Length Pile Annotation
---------
---------- ---------- -
------ -------- ----------
chr1 8538868 8539755 -
888 209 AaaaaaaaaaaaaaaaaaaA
ATREP1(100%)
chr1 9384148 9385034 +
887 231
AaaaaaaaaaaaaaaaaaaA ATREP1(100%)
chr1 18303934 18304816 +
883 1032 AaaaaaaaaaaaaaaaaaaA
ATREP1(100%)
chr2 1038042 1038914 +
873 2731 AaaaaaaaaaaaaaaaaaaA
ATREP1(100%)
chr2 2320061 2320937 - 877
2855 AaaaaaaaaaaaaaaaaaaA
ATREP1(100%)
chr4 6168349 6169230 +
882 4457 AaaaaaaaaaaaaaaaaaaA
ATREP1(100%)
chr5 23146563 23147444 +
882 5812 AaaaaaaaaaaaaaaaaaaA
ATREP1(100%)
========= ======
7 piles 881
Family
38.49
Sequence
Start End +
Length Pile Annotation
--------- ----------
---------- - ------
-------- ----------
chr1
7392255 7393392 +
1138 162 Aaaa-------------bbB VANDAL13(1%) VANDAL13(2%)
chr1
12691352 12692501 -
1150 402 Aaaaa------------bbB VANDAL13(1%) VANDAL13(1%)
chr3
18871014 18872140 -
1127 2595 Aaaa-------------bbB VANDAL13(1%) VANDAL13(1%)
chr4
10400639 10401767 +
1129 4643 Aaaa-------------bbB VANDAL13(1%) VANDAL13(2%)
========= ======
4 piles 1136
Family
44.60
Sequence
Start End +
Length Pile Annotation
--------- ----------
---------- - ------
-------- ----------
chr1
13201234 13202566 +
1333 453 --------------------
chr1
13994304 13995638 -
1335 558 --------------------
chr1
17641805 17643142 +
1338 1014 --------------------
chr1
28541147 28542484 -
1338 1346 --------------------
chr3
9171886 9173224 +
1339 1564 --------------------
========= ======
5 piles 1336
Family 0.8 is clearly an exact match to ATREP1. Family 38.49 is intriguing. The start and end of the repeat matches the terminal regions of VANDAL13, but the remainder does not. This suggests that 38.49 is a novel mobile element with terminal regions (probably LTRs) related to VANDAL13. Finally, 44.60 is not annotated by RepeatMasker, and could therefore be a novel repeat or a false positive. The relatively large family size (two larger than the minimum value of three, and hence much less likely to arise by chance or to be conserved paralogous exons) and very close agreement in length are suggestive of a novel mobile element.
PILER-PS search
PS search finds pseudo-satellites. A pseudo-satellite is a dispersed family whose members cluster in the genome (i.e., are found close together), but not so close that they form tandem arrays. The procedure is identical to PILER-DF, with one exception: hits are restricted to alignments of regions that are within a certain distance of each other (the diameter of the search). A conventional search for all self-alignments thus has a diameter of infinity (or, equivalently, the sequence length). PALS supports banded search using the -diameter parameter, for example:
pals
-self chr3.fasta -out chr3.hit -length
100 -pctid 90 -diameter 50000
See the PALS user guide for more details.
PILER-TA search
TA search finds tandem arrays. A tandem array is a
contiguous series of three or more copies of a motif. The copies are globally
alignable to each other within the chosen thresholds for local alignment.
PILER-TA searches for pyramids. A pyramid is a set of hits matching the
search criteria for belonging to a single tandem array.
Input is a GFF file containing hits annotated with pile identifiers for both images in each hit. Such a file is produced by using the -images option of the -trs command to piler.
Three GFF output files are produced, as summarized in the following table.
|
Option |
Record
type |
Description |
|
-out <tanfile> |
hit |
Contains
every hit that matches the search criteria for belonging to a pyramid. The
attribute Pyramid <n> is added to the input hit record, where <n>
is the pyramid identifier (an integer 0, 1 ...). |
|
-motif
<motiffile> |
tandemmotif |
Each record
gives the coordinates of one instance of the repeated motif found in a
pyramid. Note that only selected instances are given, the complete array may
contain many more instances of the motif. The Pyramid <n> attribute is
also given here. |
|
-pyramid
<pyrfile> |
pyramid |
Each
record gives the coordinates of a single tandem array. The PyramidIndex <n> attribute gives the identifier. |
The following figure shows a dot plot of a pyramid produced by four copies of motif B. Blue lines are hits, the red dotted line is the trivial alignment of the sequence to itself. Gray lines are the same set of hits as reflected in the lower triangle (by symmetry).
Typical commands for running PILER-TA are as follows.
piler
-trs hit.gff -images img.gff
piler
-tan img.gff -out tan.gff
-motif motif.gff -pyramid pyr.gff
For each pyramid, the sequences for each motif in motiffile can be written to a FASTA file using the –tanmotif2fasta command of piler. This works in a very similar way to the –trs2fasta command for PILER-DF. The basic command line is as follows.
piler
-tanmotif2fasta motif.gff -seq
genome.fasta
The FASTA file name is the pyramid identifier, which is an integer 0, 1 ... (N–1) for N pyramids. Options include –path, which specifies the directory (folder) path where the FASTA files should be written (default is ".", i.e. the current directory), and –prefix, which specifies a prefix to be added to the FASTA annotation. By default, the FASTA annotation is <n> <label>:<pos>, where <n> is the motif number 0, 1, ... within the pyramid, <label> is the sequence label, and <pos> is the position of the motif within the sequence.
Multiple alignments for each pyramid can be constructed using MUSCLE, and a library created from consensus sequences by following the same procedure as for PILER-DF.
The PILER-TA output files can be compared with other annotations, for example from RepeatMasker or Tandem Repeat Finder (TRF), by using the –annot command, as described above for PILER-DF. The trf2gff.py script can be used to convert a TRF .dat file to GFF format.
Cautions
We believe that PILER-TA is very specific in identifying tandem arrays—it finds few or no false positives, at least when default parameters are used. However, a single array is sometimes reported as two or more pyramids. The pyrfile output should be checked for pyramids having similar ranges (start-end coordinates), and where found they should be merged. No support is currently provided for this task in the PILER package. If this feature would be useful for your application, please check with me to see if it is now implemented; if not, I will be happy to provide scripts.
With regards to sensitivity, note that PALS alignments may not be suitable for finding arrays of short motifs, say less than 50 bases. This is partly controlled by the minimum length of a hit, as defined by the –length parameter of PALS, and also by artifacts of the implementation that would require a detailed understanding of the algorithm to explain. We recommend using Tandem Motif Finder to identify short motifs.
Note that PALS may miss some of the alignments in a pyramid.
PILER is designed to be robust against missing hits; if you do your own
processing of the output files you should be aware of this issue.
The current implementation of motif identification is preliminary and is subject to two main classes of error. (1) The motif reported by PILER is a concatenation of two or more instances of the true motif. (2) The motif is a cyclic permutation of the true motif. These are illustrated in the following diagrams. Green are the true motifs, orange the motifs reported by PILER.
PILER reports this range as the
motif.
![]()
![]()
![]()
(1) Concatenation. With a concatenation error, PILER reports n copies of the true
motif. In this case, an array of k true motifs is misidentified as an
array of k/n copies of the PILER motif.
PILER reports this range as the
motif.
![]()
![]()
![]()
(2) Cyclic permutation. With a cyclic permutation error, the boundaries
found by PILER are not optimal. In the example above, there are six true
motifs, but the PILER motif implies seven instances, two of which are fragments
(cross-hatched). By parsimony, the true motif requires fewer mutational events
and therefore better suggests the history of this array.
These issues with motif identification are relatively unimportant for repeat masking, but should be considered if the goal is an understanding of evolutionary mechanisms and history. Improved motif identification is an active area of research—by the time you read this, we may have better methods, so feel free to contact me to ask.
Optional parameters
There are three algorithm parameters for the –tan
command. They are briefly summarized in the following table.
|
Option |
Description |
|
-minhitcount n |
The minimum number of hits required to infer a
pyramid. Default n=2. Must have n > 1. |
|
-maxmargin m |
Floating point value specifying how well hit
endpoints must align, as a fraction of the hit length. Default m=0.05.
Should have 0 <~ m << 1. |
|
-minratio r |
Floating
point value specifying the minimum length ratio required to infer that a pair
of hits that belong to the same pyramid. Default r=0.5. Should have r
≥ 0.5. |
For further explanation of these parameters, please refer to the paper (if one is published), or contact me. The ‑minhitcount and –minratio values should probably be left as their defaults. Possibly, increasing –minhitcount might improve specificity if you get false positives. The ‑maxmargin (m) parameter is the one that is most likely to be useful. Smaller values increases the stringency of the search criteria, larger values make the search looser. So smaller values of m might improve specificity if you are getting false positives, larger values of m might improve sensitivity if you are getting false negatives.
PILER-TR
TR search finds sets of pairs of repeats that may be related terminal repeats due to mobile elements such as the LINE-LTR superfamilies or Tc1.
Input is a set of hits, as for PILER-DF and -PS.
Output is a set of families, defined in a similar way to PILER-DF and -PS.
The command line is:
piler
-tr hits.gff -out tr.gff
The output file is in GFF format, feature is "tr". Each record gives the range of a single pair of terminal repeats, from the start of the first repeat to the end of the second repeat in the pair, and is assigned to a family. A typical record is as follows.
chr5 piler tr 12832560 12833058 0 + . Family 12 ; Cand
8158
The Family attribute gives the family and the Cand attribute is a reference to the candidate output file, which is specified by the -cand <filename> option and contains a list of all candidate pairs found in the first pass of the search.
The TR search algorithm
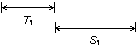
The first phase searches for candidate pairs of terminal repeats. A candidate pair is a single hit. A candidate hit is recognized length constraints on the repeats (i.e., the images in the hit) and the distance between them.
![]()

A
candidate pair of terminal repeats. The repeat length T and the spacing S
(distance between the end of the first repeat and start of the second) have
length constraints (minimum and maximum) that are parameters of the search
algorithm.
The second phase searches for pairs of candidates that confirm each other. Pairs confirm each other if (a) the terminal repeats from the two pairs align globally to each other, and (b) the spacing between them is similar. Confirming pairs are recognized by finding hits that align one repeat from each pair.
![]()
![]()
![]()
![]()
![]()

![]()
A mutually confirming pair of candidates. Hits hit1 and hit2
are identified as candidates in the first
pass. The second pass searches for hits such as hit3 that
join a pair of candidates which confirm each other. A large value of D
is used in an attempt to minimize false positives due to satellites and
pseudo-satellites.
The third phase finds families of confirming pairs. The minimum family size is 2, meaning that just one hit like hit3 is sufficient.
Parameters of the search algorithm are summarized in the following table, referring to the above diagrams.
|
Option |
Description |
|
-mintrlength |
Phase 1. Minimum value of T. Default 50. |
|
-maxtrlength |
Phase 1. Maximum value of T. Default 2000. |
|
-mintrspacing |
Phase 1. Minimum value of S. Default 50. |
|
-maxtrspacing |
Phase 1. Maximum value of S. Default 12000. |
|
-minspacingratio |
Phase 2. Floating point. Minimum value of min(S1,
S2) / max(S1, S2).
Default 0.9. |
|
-minhitratio |
Phase 2. Minimum value of min(T1,
T2) / max(T1, T2).
Default 0.5. |
|
-mindistpairs |
Phase 2. Minimum value of D. Default 50,000. A large value of D is recommended in order to minimize false
positives due to satellites and pseudo-satellites. |
|
-minfam |
Phase 3. Minimum family size. Default 3. |
Caution
While we have found PILER-TR to be useful in identifying novel families of mobile elements, we feel that it is not yet a mature tool. In contrast to the other PILER search methods, we have not yet found an algorithm design and parameter values that offer high specificity and sensitivity. We therefore emphasize that PILER-TR is a tool that provides candidates for further analysis. This is of course also true of the other PILER search methods. Improving PILER-TR is an active area of research.